Birth defects
Birth defects are inborn errors of development, which include any structural or functional anomaly with effects on physical, intellectual, and social wellbeing [1]. Birth defects represent a considerable and increasing clinical and public health challenge due to their worldwide impact on population health. Major birth defects are common, costly, and critical [2]. Collectively, they occur in 4-8 in 100 live births worldwide [3, 4], which translates into an estimated 8.5 million affected babies each year. In the US alone, the cost of care during a single year is estimated at $3 billion [5], along with considerable indirect and lifelong personal and societal costs. Finally, many birth defects critically affect survival. In the US, birth defects are the leading cause of infant mortality [6]; they are present in one in every 5 deaths in the first year of life, and are associated with thousands of deaths each year.
Identifying a concerning genetic finding during pregnancy does not necessarily mean a medical recommendation to terminate the pregnancy. In up to 70% of cases birth defects can be prevented, but also affected children can be offered care that could be life saving, or would reduce the severity of disability via early intervention strategies. These intervention strategies and treatments are relevant after birth but also, and especially so, in early pregnancy. The United States and other high-income countries reported a remarkable 46 percent decline in infant mortality rates from birth defects over the period 1980 to 2001, and much of this reduction can be attributed to improvements in diagnosis, care and prevention [4].
Prenatal diagnosis
Detection of fetal anomalies during pregnancy is referred to as prenatal diagnosis. It is a broad field that integrates several medical areas, from obstetrics and gynecology to genetics and pediatrics. Diagnosis of fetal diseases during pregnancy is performed using several approaches, typically ultrasound and blood markers. Abnormal findings or measurements in these two methods are used for statistical risk assessment, and they are usually not considered as diagnostic, i.e., verification through an invasive method is required.
Invasive methods for prenatal diagnosis include amniocentesis (amniotic fluid test), and chorionic villus sampling (CVS). Both invasive methods carry a risk of miscarriage or other complications. Due to several clinical reasons, invasive methods are limited to specific time windows during pregnancy: amniocentesis is performed at 15-20 weeks, or later at 30-32 weeks; CVS is performed at 10-13 weeks of gestation, and is suggested as the test of choice in the first trimester.
Different molecular methods are used to analyze the DNA obtained through the invasive methods. Structural mutations, which are large parts of the genome that are missing or duplicated, are detected using karyotyping, fluorescent in situ hybridization (FISH) or comparative genomic hybridization (CGH). Structural mutations can be at the level of a complete chromosome (e.g., Down syndrome), or smaller, sub-chromosomal aberrations (e.g., DiGeroge syndrome). Point mutations, which are single nucleotides (DNA building blocks) that were changed, along with indels (several-to-tens of nucleotides that are inserted or deleted), are detected using next generation sequencing (NGS). NGS enables whole genome sequencing (WGS), whole exome sequencing (WES), or sequencing of panels that contain specific regions of interest in the genome; it is usually performed over a trio, i.e., both the parents and the fetus.
Noninvasive prenatal diagnosis
During the last decade, noninvasive prenatal tests (NIPT) have emerged as a simple, risk-free, and accurate method to detect various fetal anomalies. NIPT is based on the presence of DNA fragments that circulate in our blood plasma, outside of blood cells, named cell-free DNA (cfDNA). In pregnant women, the cfDNA consists of both maternal and fetal cfDNA. The number of fetal fragments out of all cfDNA fragments is termed “fetal fraction”, and reaches approximately 10% near the end of the first trimester.
The first clinical uses of NIPT were for chromosomal-level anomalies (a.k.a. aneuploidies). The most notable example is NIPT for trisomy 21 (where the genome of the fetus contains an extra copy of chromosome 21), which causes Down syndrome, a test that has shown high sensitivity and specificity not only in high-risk populations like advanced maternal age pregnancies, but also in the general population [7]. Trisomies 13 and 18 are tested today as well, with high specificity. Another chromosome-related clinical application is fetal sex determination, which also enables to rule out X-linked recessive disorders in case of a female fetus.
Recently, NIPT has also become available for large sub-chromosomal variations, such as microdeletions/microduplications. This is important given that the aneuploidy incidence in pregnancy is 1-2%, whereas the collective incidence of microdeletions/microduplications is 3.6% [8, 9].
Today, NIPT is still not available for point mutations and small insertions-deletions. Out of all birth defects that have a known genetic cause, single gene diseases account for up to 30% of the cases (Figure 1) [10]. Out of the birth defects with an unknown cause, an increasing number is found in the last decade to have a genetic cause that can be revealed only with modern diagnosis technologies [11]. It should be noted that some severe single-gene disorders that are prenatally tested with invasive methods have an adult-onset and are therefore not included in the above statistics (as they do not cause birth defects).
It is generally accepted that prenatal diagnosis will eventually be almost entirely noninvasive. NIPT will not only replace invasive methods in many high-risk pregnancies but will also be used as a screening test in the entire population, which otherwise mostly avoids the invasive alternatives. Since NIPT currently covers only a handful of anomalies (several chromosomal and large sub-chromosomal aberrations), the main challenge in getting to a complete noninvasive solution remains point mutations and small insertions-deletions (from 1 bp to tens-of-thousands) [12]. This is exactly IdentifAI’s specialty.
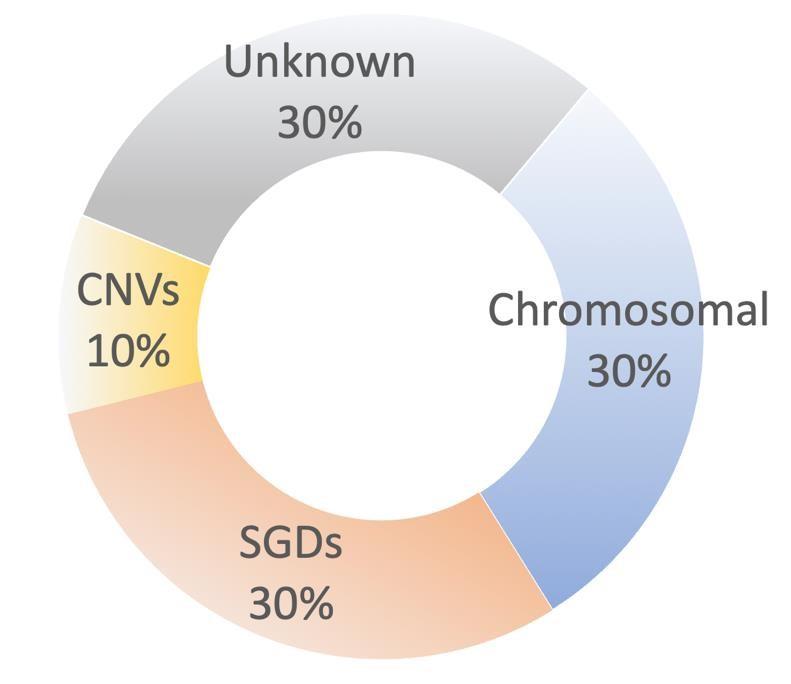
Figure 1. Out of all genetic birth defects (with the exception of adult-onset conditions that are prenatally tested, which are not shown here), an estimated 30% are a result of chromosomal abnormalities, which are only partially detected with current NIPT approaches. Up to 10% of genetic birth defects are estimated to be a result of insertions-deletions of varying sizes, which cause copy number variations (CNV) and can be detected by chromosomal microarray (CMA). Most cases (an estimated 60%) are undetected using current NIPT solutions, and up to 50% of these cases are estimated to be a result of point mutations that lead to single gene disorders (SGD) [10].
Identifai’s solution
IdentifAI’s solution (US patent US20210340601A1 [13]) offers an early, risk-free, “one-stop shop” for all types of mutations, i.e., chromosomal, sub-chromosomal, and meaningful point mutations.
As many prenatal diagnosis methods are available, and their advantages and disadvantages vary, choosing the right test or tests becomes challenging. Most importantly, prenatal tests differ in diagnostic yield, i.e., how many possible anomalies are covered. Different tests also have varying risks and complications, costs, convenience levels and availability during pregnancy. CMA, for example, covers chromosomal and subchromosomal anomalies, but it is invasive, and therefore carries a substantial risk to the pregnancy. Chromosomal NIPT offered today is risk-free, but covers only a small number of anomalies, thus the diagnostic yield is low. In many cases, when faced with the large amount of incomplete available solutions, expectant couples experience “decision fatigue”, leading to a decline in their ability to make optimal decisions about the pregnancy.
The ideal test should thus be available early during pregnancy, and in every stage of it; risk-free, i.e., noninvasive; have a high diagnostic yield, even in low-risk population; easily updated based on up-todate gene and disease databases; offer clear and meaningful findings, i.e., only pathogenic mutations, causing meaningful pathologies; affordable and cost-effective compared to alternatives; and convenient – a “one-stop shop”, that is available anywhere.
Developing a deep and accurate sequencing protocol
IdentifAI‘s approach begins with blood samples drawn from the parents. The maternal and paternal DNA is extracted from white blood cells. The cfDNA, containing both fetal and maternal DNA, is extracted from the plasma. All DNA samples are sequenced using WGS. The cfDNA sample is sequenced to a high depth of 300, meaning that each genomic position is read independently 300 times. To decipher the fetal genome, the genetic information from the parents and the cfDNA is analyzed by a proprietary algorithm named Hoobari [14, 15].
Different types of point mutations require different solutions
The various types of potentially harmful genetic variations pose a substantial algorithmic challenge. We tackle this challenge by dividing the problem into smaller ones, based on our expertise in the genetic and medical domains. Point mutations cause Mendelian disorders, also called monogenic diseases. These disorders are caused by a single gene and follow Mendelian inheritance, and are divided into autosomal recessive (AR) and autosomal dominant (AD) diseases. In AR diseases, two copies of a mutated gene (either a mutation in the very same nucleotide, or a mutation elsewhere in the same gene) were inherited, one from each parent. If different mutations in the same gene are inherited, this is called “compound heterozygosity”, and if the same mutation is inherited, it is called “homozygous mutation”. In AD diseases, inheriting one copy of a mutated gene is enough to cause a disease. Other mendelian diseases are caused by mutations in sex chromosomes, mostly on chromosome X (termed X-linked). Finally, some mendelian diseases are caused by de novo mutations, that spontaneously occur during gametogenesis (the creation of sperm and egg cells) and are found in the fetus but not in the parents. The different inheritance patterns of single gene disorders are based on inheriting mutations from the mother, from the father or from both parents, and these options require different computational solutions.
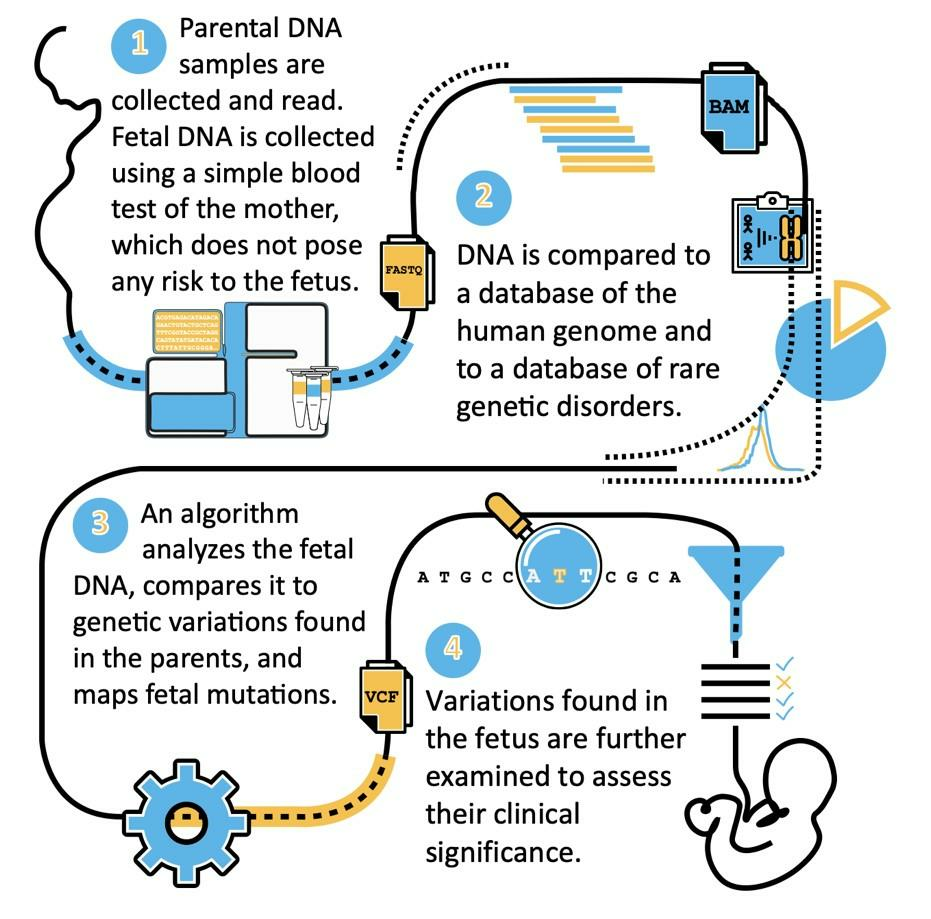
Figure 2. The general workflow of IdentifAI’s approach. During steps 2-3, our proprietary algorithm creates a fetal-maternal DNA classifier and uses it together with genetic information from both parental genomes to detect fetal mutations.
Detection of point mutations
Cases where only the father has a mutation are solved by looking for presence of DNA fragments that support the paternal mutation in the maternal blood. This enables detecting AD diseases of paternal origin, and compound heterozygous AR diseases. A similar solution is used for de novo mutations. Cases in which the mother carries a mutation (whether the father carries it or not) are solved with more sophisticated algorithms, which are based on imbalances between the normal copy and the mutation within the cfDNA. If the mutation is over-represented, there is a higher likelihood of it being inherited by the fetus. To detect mild imbalances, our algorithm assesses each DNA fragment separately and searches for maternal and fetal features and signatures. Using this high-resolution method, we manage to accurately detect the two most computationally challenging categories of disease-causing point mutations: AD maternally derived diseases and homozygous mutation AR diseases, where the same mutation exists in both parents.
Analyzing genomes using domain knowledge, statistics, and AI
IdentifAI‘s approach originated in academia, in a laboratory that specialized for years in NGS analysis and genomics, and detection of Mendelian diseases (the lab’s methods have also lead to industrial solutions and have spawned biotech companies). Using this knowledge and experience, we understood that NIPT of point mutations requires a variant caller, i.e., a bioinformatics algorithm that separately assesses each DNA fragment, to detect mutations. Today, our solution consists of several statistical and AI algorithms, that are all in advanced stages of several patent submissions. In our method, we use an AI-based approach; we develop a fetal-maternal cell-free DNA classifier, i.e., a machine-learning method that predicts whether a fragment is most likely derived from fetal or maternal origins. Then, inspired by similar software developed by leading laboratories (e.g., MIT and Harvard’s Broad Institute’s Genome Analysis Toolkit, and Sanger institute’s Samtools), we developed a Bayesian algorithm that intelligently integrates all the evidence at each genomic position to detect a mutation. Instead of using different methods to filter-out uninformative DNA fragments, we use soft-scoring for each fragment, thus utilizing all available information.
Furthermore, after assessing each potential variant independently, we incorporate another layer of information, utilizing nearby variants to improve prediction accuracy. Specifically, we rely on genetic linkage and haplotypes, i.e., genomic loci that are physically proximate on the same copy of the chromosome (either maternally- or paternally-derived) and tend to be inherited together. Our algorithm assembles haplotypes based on overlapping DNA fragments and uses these haplotypes to improve upon our genotype predictions.
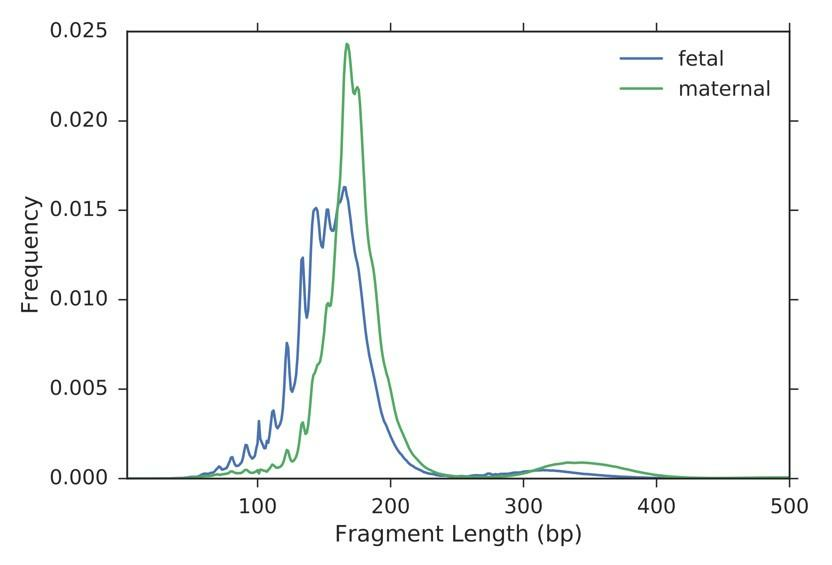 Figure 3. Fetal and maternal cell-free DNA in the maternal plasma have different fragment length distributions. This is an example of a feature that is used in the fetal-maternal cellfree DNA fragment classifier.
Figure 3. Fetal and maternal cell-free DNA in the maternal plasma have different fragment length distributions. This is an example of a feature that is used in the fetal-maternal cellfree DNA fragment classifier.
Finally, we leverage even more available information: our method is able to learn from previous analyses and our unique, ever-growing database of real-family data, and keeps improving with time. This is achieved by an additional AI-based approach, which utilizes information that may often be overlooked by the Bayesian algorithm. This approach is also inspired by widely accepted solutions to similar problems, that are used today in the field of genome analysis.
The above methods (and others) enable the complete coverage of point mutations across the genome, using a custom-fit solution for each challenge in the NIPT of point mutations. By solving this missing part of the puzzle, we managed to develop a risk-free, early, up-to-date technology, that covers all genetic disorders across the genome and enables a convenient, affordable, and cost-effective test.
Results
First, we present the accuracy of our core Bayesian algorithm.
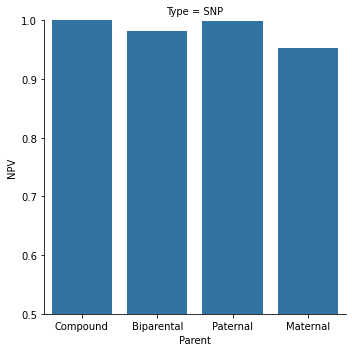
Figure 4. Our capabilities in an 11 weeks gestation family are shown here, for 4 different categories of point mutations. Paternal and compound heterozygous mutations are the more common cases, while maternal and biparental (homozygous fetal mutation) are rarer.
Second, since we practically solved the more common Compound heterozygous and Paternal categories, we aimed to improve the Maternal and Biparental accuracies. Using our AI-based prediction correction and then predicting the inherited haplotypes, we dramatically improved our accuracy in these categories
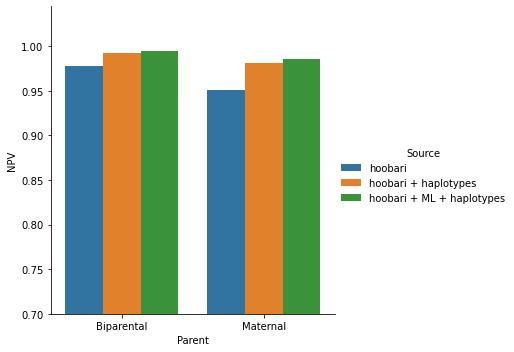
Figure 5. Our capabilities in an 11 weeks gestation family are shown here, for maternal and biparental categories, before and after adding AI-based correction and haplotypes information.
Out of 100 fetuses predicted as healthy –
In the “Compound” category, 100 will actually be healthy, and 0 are expected false negatives
In the “Paternal” category, 99.8 will actually be healthy, and 0.2 are expected false negatives
In the “Biparental” category, 99.5 will actually be healthy, and 0.5 are expected false negatives
In the “Maternal” category, 98.5 will actually be healthy, and 1.5 are expected false negatives
References
- World Health Assembly 63. World Health Assembly, 63. (2010). Birth defects: report by the Secretariat. World Health Organization. World Health Organization; 2010.
- Feldkamp ML, Carey JC, Byrne JLB, Krikov S, Botto LD. Etiology and clinical presentation of birth defects: population based study. BMJ. 2017;357:j2249.
- Centers for Disease Control and Prevention (CDC). Update on overall prevalence of major birth defects–Atlanta, Georgia, 1978-2005. MMWR Morb Mortal Wkly Rep. 2008;57:1–5.
- Christianson A, Howson CP, Modell B. Christianson A, Howson CP, Modell B. March of Dimes Global Report on Birth Defects.The Hidden Toll of Dying and Disabled Children. March of Dimes Birth Defects Foundation, 2006. March Dimes Glob Rep Birth Defects Hidden Toll Dying Disabl Child. 2005.
- Russo CA, Elixhauser A. Hospitalizations for Birth Defects, 2004: Statistical Brief #24. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville (MD): Agency for Healthcare Research and Quality (US); 2006.
- Matthews TJ, MacDorman MF, Thoma ME. Infant Mortality Statistics From the 2013 Period Linked Birth/Infant Death Data Set. Natl Vital Stat Rep Cent Dis Control Prev Natl Cent Health Stat Natl Vital Stat Syst. 2015;64:1–30.
- van der Meij KRM, Sistermans EA, Macville MVE, Stevens SJC, Bax CJ, Bekker MN, et al. TRIDENT-2: National Implementation of Genome-wide Non-invasive Prenatal Testing as a First-Tier Screening Test in the Netherlands. Am J Hum Genet. 2019;105:1091–101.
- Hillman SC, Pretlove S, Coomarasamy A, McMullan DJ, Davison EV, Maher ER, et al. Additional information from array comparative genomic hybridization technology over conventional karyotyping in prenatal diagnosis: a systematic review and meta-analysis. Ultrasound Obstet Gynecol Off J Int Soc Ultrasound Obstet Gynecol. 2011;37:6–14.
- Shaffer LG, Dabell MP, Fisher AJ, Coppinger J, Bandholz AM, Ellison JW, et al. Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat Diagn. 2012;32:976–85.
- Vossaert L, Zemet R, Van den Veyver IB. Advances in Non-Invasive Diagnosis of Single-Gene Disorders and Fetal Exome Sequencing. Handb Genet Diagn Technol Reprod Med Improv Patient Success Rates Infant Health. 2022;:301.
- Basel-Salmon L, Orenstein N, Markus-Bustani K, Ruhrman-Shahar N, Kilim Y, Magal N, et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet Med. 2019;21:1443–51.
- Rabinowitz T, Shomron N. Genome-wide noninvasive prenatal diagnosis of monogenic disorders: Current and future trends. Comput Struct Biotechnol J. 2020;18:2463–70.
- Shomron N, RABINOWITZ T. Method and system for identifying gene disorder in maternal blood. 2021.
- Rabinowitz T, Polsky A, Golan D, Danilevsky A, Shapira G, Raff C, et al. Bayesian-based noninvasive prenatal diagnosis of single-gene disorders. Genome Res. 2019. https://doi.org/10.1101/gr.235796.118.
- Rabinowitz T, Deri-Rozov S, Shomron N. Improved noninvasive fetal variant calling using standardized benchmarking approaches. Comput Struct Biotechnol J. 2021;19:509–17.